The first chapter of my PhD Thesis is published! For this project I inherited a dataset produced before I joined the lab. The data consisted of a large RNA-Seq dataset for three Macrostomum species (M. hystrix, M. spirale and M. pusillum) with high sequencing depth across multiple biological conditions. We had RNA-Seq for adults, juveniles and regenerants. Regenerants are a mixture of animals collected at increasing time post amputation of the tail. It was clear from the beginning that we wanted to use annotations from M. lignano to annotate groups of orthologous genes and then look at rates of evolution and gene expression. However, since the data was collected with the primary aim to produce high-quality transcriptomes, some of the biological conditions were not comparable between species. Specifically, we found that the M. pusillum juveniles were already quite advanced in development making the Hatchling - Adult contrast tricky to compare across the genus. You can see this in the picture below where gene expression between hatchlings and adults are compared. In both M. hystrix and M. spirale there is a large cloud to the right of the diagonal line representing transcripts with higher expression in adults, but in M. pusillum this cloud is shifted closer to the diagonal.
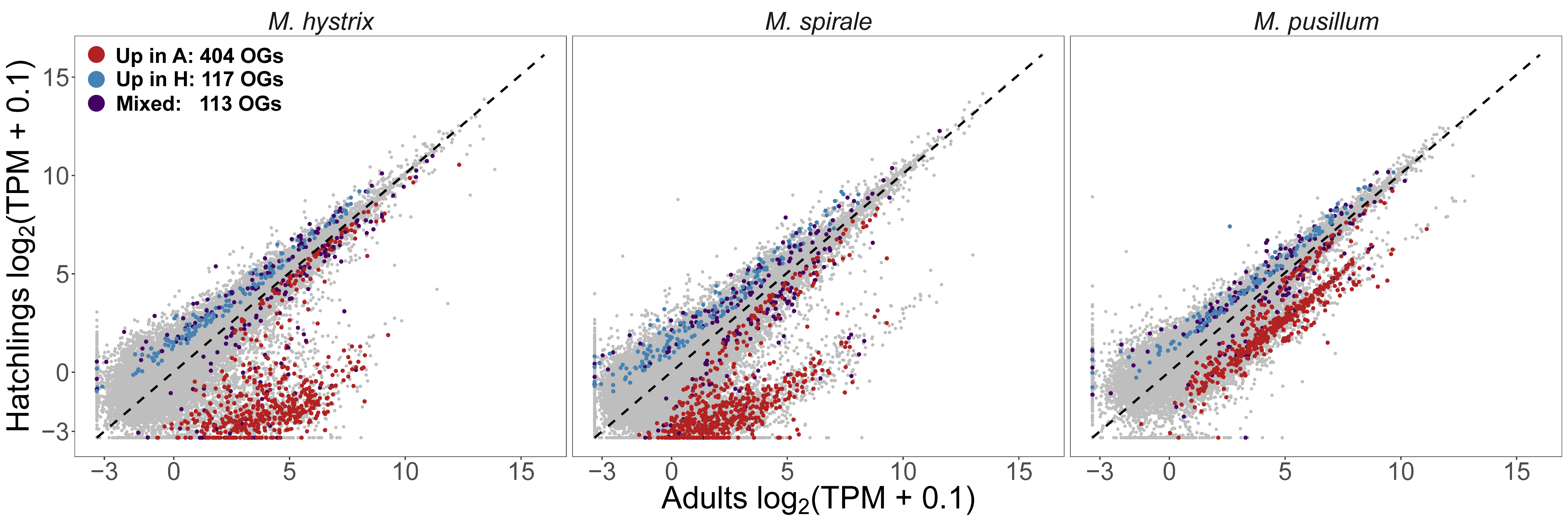
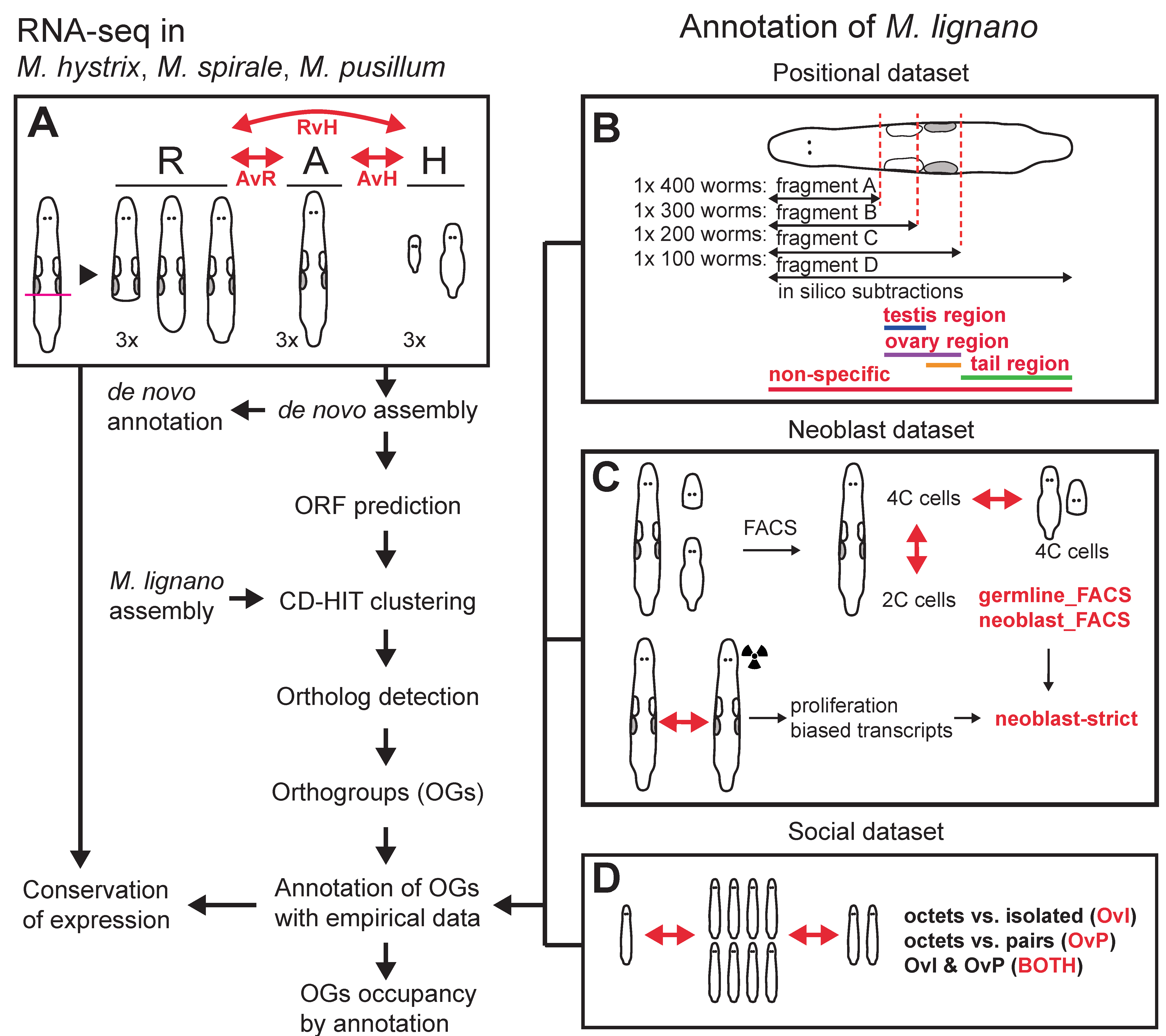
Because of this, we had to abandon our analysis of gene expression evolution and focused on sequence-based measures and homology detection failure. We transferred annotations from M. lignano to the other species, which allowed us to identify reproduction-related genes. To transfer the annotations we reannotated the most up-to-date transcriptome of M. lignano with previous RNA-Seq experiments. You can see a schematic of our workflow below. You might rightfully think that this looks quite complicated. Indeed, making this reannotation was hard work that is now mostly hidden in the Supplementary material… Axel Wiberg was very helpful in getting this done.
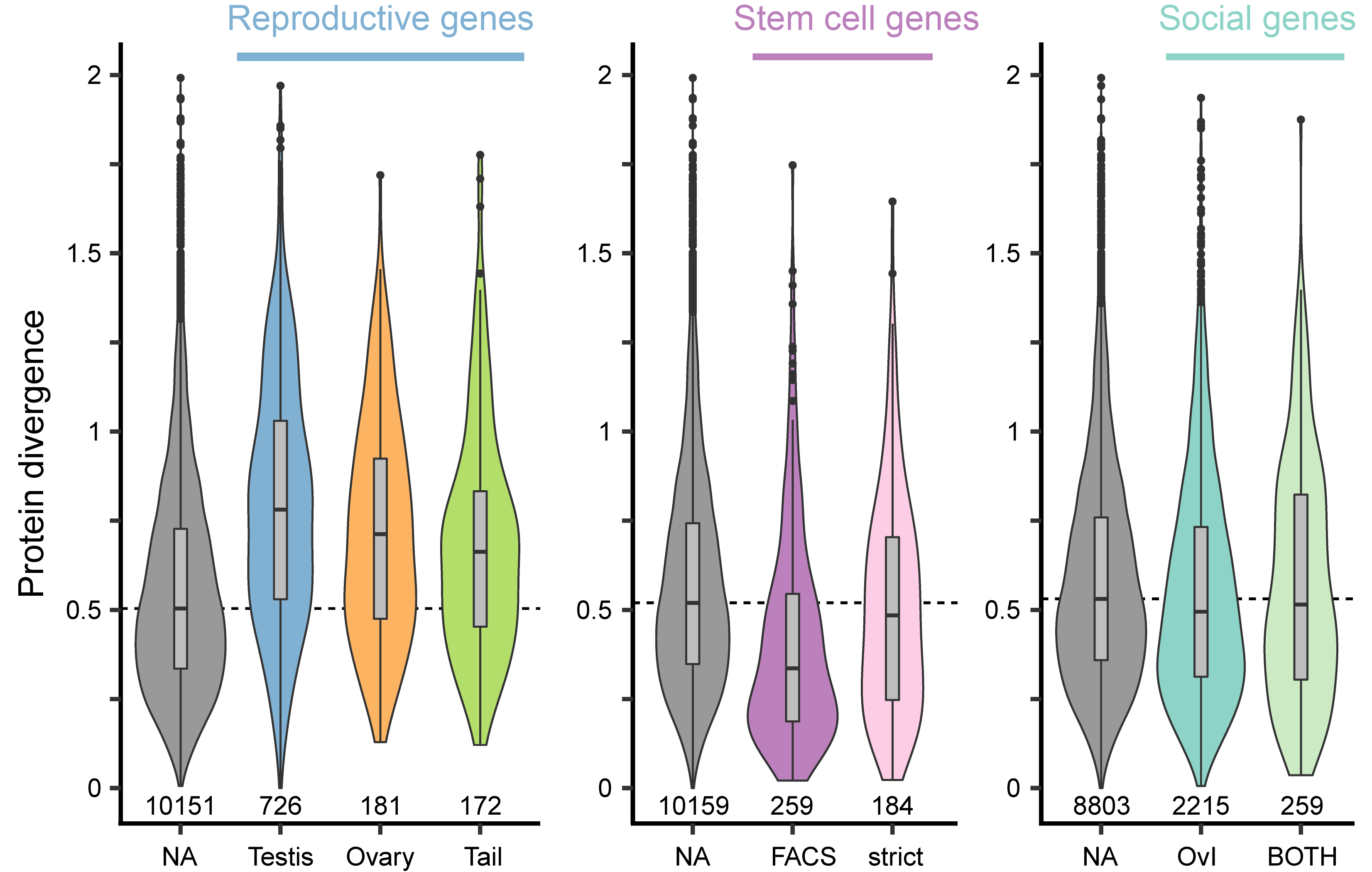
Due to male-female coevolution genes should diverge rapidly but this has not been looked at in flatworms. We could show that reproduction-related genes have a higher sequence divergence and are also harder to detect with phylogenetic distance. Below you see a summary of sequence divergence across the genus of proteins with different annotations. We can see that reproductive genes have a higher divergence compared to the grey background genes. Further, stem cell genes are more conserved, while genes that were sensitive to social condition were not different from the background.
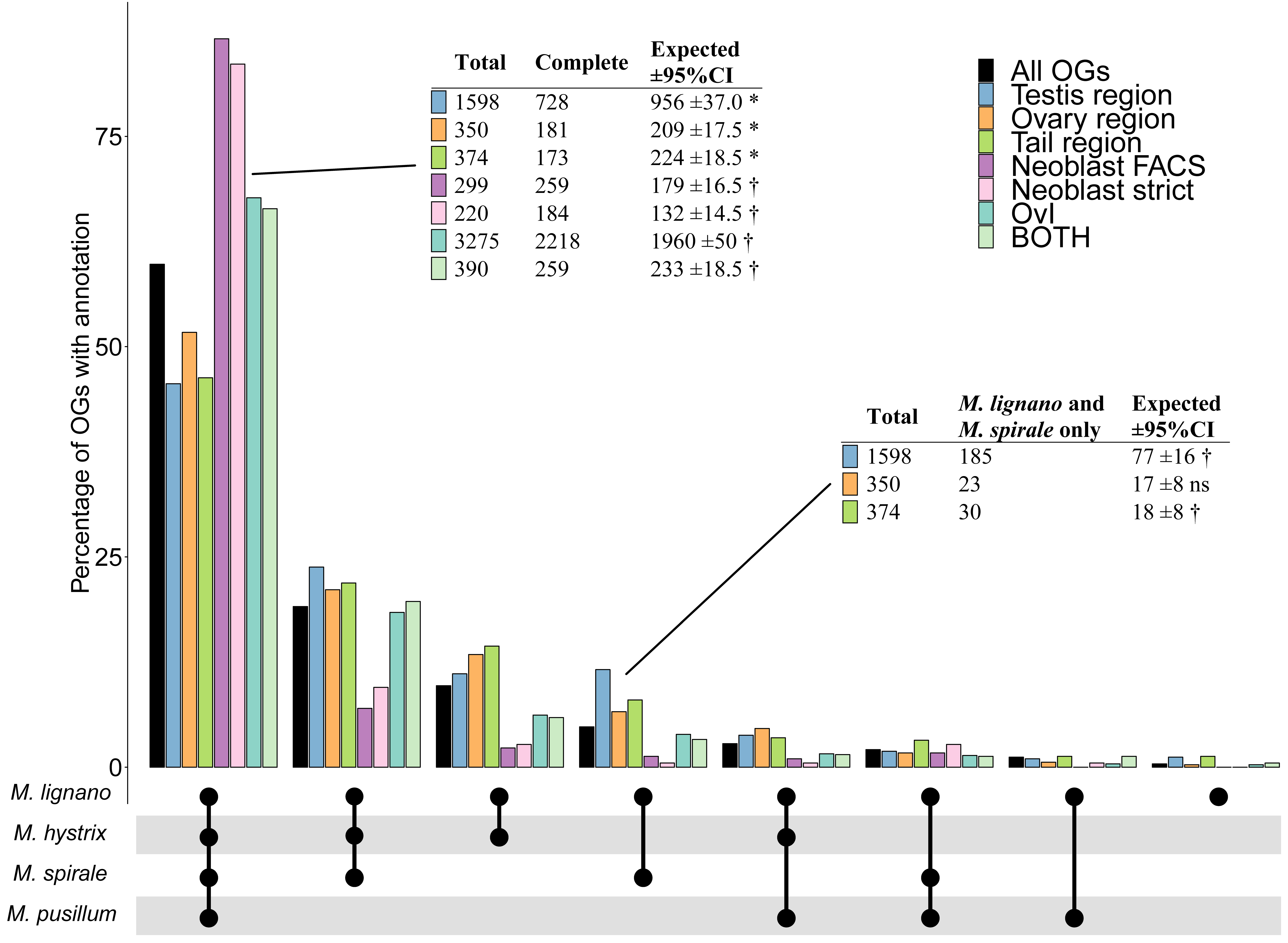
We find a similar story when looking at homology detection failure. Orthogroups with a stem cell annotation are more often complete (contain all four species) than a random selection of genes. Here orthogroups with reproduction-related genes are less often complete than expected, presumably because they have diverged in sequence to the point that homology could not be detected.
In summary, we can say that sexual selection likely also drives rapid evolution of these genes in flatworms. I am delighted we could find such an exciting result given the initial trouble with the data. You can read the paper here if you want to learn more about this.